Get started¶
Let us get a latest stable version of flowr, from CRAN. To get the latest features, your could also try the version from github.
install.packages('flowr') ## CRAN
install.packages('devtools')
devtools::install_github("sahilseth/flowr")
We have a quite handy command-line-interface for flowr, which exposes all functions of the package to terminal. Such that we dont have to open a interactive R session each time. To make this work, run a setup function which copies the ‘flowr’ helper script to your ~/bin directory. If you would like to do a test drive on its other capabilities, here are a few examples.
library(flowr)
setup()
## You could try this from the terminal
Rscript -e 'library(flowr);setup()'
## now run the following to confirm that ~/bin is added your PATH variable:
echo $PATH
## if ~/bin is not in your path, run and add the following to your ~/.bashrc
export PATH=$PATH:~/bin
## now run the following from the terminal, to check if setup worked fine.
flowr
Toy example¶

Consider, a simple example where we have three instances of the sleep command running ( which basically stalls the terminal for few seconds and does nothing ). After its completion three tmp files are created with some random data. After this, a merge step follows, which combines them into one big file. Next we use du to calculate the size of the resulting file. This flow is shown in the above described figure.
NGS context This is quite similar in structure to a typical workflow from where a series of alignment and sorting steps may take place on the raw fastq files. Followed by merging of the resulting bam files into one large file per-sample and further downstream processing.
To create this flow in flowr, we need the actual commands to run; and some kind of a configuration file to describe which ones go first.
Here is a table with the commands we would like to run ( or flow mat ).
| samplename | jobname | cmd |
|---|---|---|
| sample1 | sleep | sleep 10 && sleep 2;echo hello |
| sample1 | sleep | sleep 11 && sleep 8;echo hello |
| sample1 | sleep | sleep 11 && sleep 17;echo hello |
| sample1 | create_tmp | head -c 100000 /dev/urandom > sample1_tmp_1 |
| sample1 | create_tmp | head -c 100000 /dev/urandom > sample1_tmp_2 |
| sample1 | create_tmp | head -c 100000 /dev/urandom > sample1_tmp_3 |
| sample1 | merge | cat sample1_tmp_1 sample1_tmp_2 sample1_tmp_3 > sample1_merged |
| sample1 | size | du -sh sample1_merged; echo MY shell: $SHELL |
Further, we use an additional file specifying the relationship between the steps, and also other resource requirements: flow_def. Each row in a flow mat relates to one job.
Notice how jobname column is being used a key throught the two tables. And how prev_jobs (previous jobs) defines what jobs need to complete before the one described in that row starts.
| jobname | sub_type | prev_jobs | dep_type | queue | memory_reserved | walltime | cpu_reserved | platform | jobid |
|---|---|---|---|---|---|---|---|---|---|
| sleep | scatter | none | none | short | 2000 | 1:00 | 1 | torque | 1 |
| create_tmp | scatter | sleep | serial | short | 2000 | 1:00 | 1 | torque | 2 |
| merge | serial | create_tmp | gather | short | 2000 | 1:00 | 1 | torque | 3 |
| size | serial | merge | serial | short | 2000 | 1:00 | 1 | torque | 4 |
Stitch it¶
We use the two files descirbed above and stich them to create a flow object, which contains all the information we need for submission to the cluster. Additionally we can give a name to this flow, using flowname argument and also override the platform described in flow def. Look at to_flow help file for more information.
fobj <- to_flow(x = flow_mat, def = as.flowdef(flow_def),
flowname = "example1", platform = "lsf")
Plot it¶
We can use plot_flow to quickly visualize the flow; this really helps when developing complex workflows. Additionally, this function also works on the flow definition table as well (plot_flow(flow_def).
plot_flow(fobj) # ?plot_flow for more information
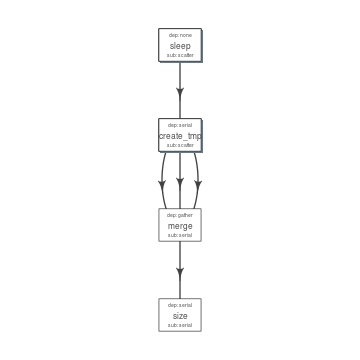
Flow chart describing process for example 1
Dry Run¶
Dry run: Quickly perform a dry run, of the submission step. This creates all the folder and files, and skips submission to the cluster. User’s may spend some time checking the *.sh files for each of the jobs along with pdf of the flow etc.
submit_flow(fobj)
Test Successful!
You may check this folder for consistency. Also you may re-run submit with execute=TRUE
~/flowr/type1-20150520-15-18-27-5mSd32G0
Submit it¶
Submit to the cluster !
submit_flow(fobj, execute = TRUE)
Flow has been submitted. Track it from terminal using:
flowr::status(x="~/flowr/type1-20150520-15-18-46-sySOzZnE")
OR
flowr status x=~/flowr/type1-20150520-15-18-46-sySOzZnE
Check its status¶
One may periodically run status to monitor the status of a flow.
Note: Please make sure to include x=~ in status, to expicitly define the variable. Also unlike other command line tools you may skip adding “-” in from of each argument ( no need of -x=~).
flowr status x=~/flowr/type1-20150520-15-18-46-sySOzZnE
Showing status of: /rsrch2/iacs/iacs_dep/sseth/flowr/type1-20150520-15-18-46-sySOzZnE
| | total| started| completed| exit_status| status|
|:---------|-----:|-------:|---------:|-----------:|---------:|
|001.sleep | 10| 10| 10| 0| completed|
|002.tmp | 10| 10| 10| 0| completed|
|003.merge | 1| 1| 1| 0| completed|
|004.size | 1| 1| 1| 0| completed|
Alternatively, to check a summarized status of several flows, skip the full path, and mention only the parent direcotry, for example:
flowr status x=~/flowr/type1-20150520-15-18-46-sySOzZnE
Showing status of: /rsrch2/iacs/iacs_dep/sseth/flowr/type1-20150520-15-18-46-sySOzZnE
| | total| started| completed| exit_status| status|
|:---------|-----:|-------:|---------:|-----------:|---------:|
|001.sleep | 30| 30| 10| 0|processing|
|002.tmp | 30| 30| 10| 0|processing|
|003.merge | 3| 3| 1| 0| pending|
|004.size | 3| 3| 1| 0| pending|
Scalability: Quickly submit, and check a summarized OR detailed status on ten or hundreds of flows.
Kill it¶
Incase something goes wrong, one may use to kill command to terminate all the relating jobs.
kill one flow:
flowr kill_flow x=flow_wd
One may instruct flowr to kill multiple flows, but flowr would confirm before killing.
kill(x='fastq_haplotyper*')
Flowr: streamlining workflows
found multiple wds:
./fastq_haplotyper-MS132-20150825-16-24-04-0Lv1PbpI
/fastq_haplotyper-MS132-20150825-17-47-52-5vFIkrMD
Really kill all of them ? kill again with force=TRUE
To kill multiple, set force=TRUE:
kill(x='fastq_haplotyper*', force = TRUE)
While submission is in progress, and you figure, you want to kill the flow; its best to let submit_flow do its job, when done simply use kill(flow_wd). If submit_flow is interrupted, files with details regarding job ids etc are not created, thus flowr can’t associate submitted jobs with flow instance ( hence can’t kill them ). In such a situation you may resort to killing them manually.
## manual killing:
jobids=$(qstat | grep 'mypattern')
qdel $jobids
Re-run a flow¶
flowr also enables you to re-run a pipeline in case of hardware or software failures.
- hardware failure: no change to the pipeline is required, simply rerun it:
rerun(x=flow_wd, start_from=<intermediate step>) - software failure: either a change to flowmat or flowdef has been made:
rerun(x=flow_wd, mat = new_flowmat, def = new_flowdef, start_from=<intermediate step>)
In either case there are two things which are always required, a flow_wd (the folder created by flowr which contains execution logs) and name of the step from where we want to start execution. Refer to the help section for more details.
Ingredients for building a pipeline¶
An easy and quick way to build a workflow is create to create a set of two tab delimited files. First is a table with commands to run (for each module of the pipeline), while second has details regarding how the modules are stitched together. In the rest of this document we would refer to them as flow_mat and flow_def respectively (as introduces in the above sections).
Both these files have ajobnamecolumn which is used as a ID to connect them to each other.
We could read in, examples of both of these files to understand their structure.
## ------ load some example data
ex = file.path(system.file(package = "flowr"), "pipelines")
flow_mat = as.flowmat(file.path(ex, "sleep_pipe.tsv"))
flow_def = as.flowdef(file.path(ex, "sleep_pipe.def"))
1. Flow Definition¶
Each row in this table refers to one step of the pipeline. It describes the resources used by the step and also its relationship with other steps, especially, the step immediately prior to it.
It is a tab separated file, with a minimum of 4 columns:
jobname: Name of the stepsub_type: Short for submission type, refers to, how should multiple commands of this step be submitted. Possible values areserialorscatter.prev_job: Short for previous job, this would be jobname of the previous job. This can be NA/./none if this is a independent/initial step, and no previous step is required for this to start.dep_type: Short for dependency type, refers to the relationship of this job with the one defined inprev_job. This can take valuesnone,gather,serialorburst.
These would be explained in detail, below.
Apart from the above described variables, several others defining the resource requirements of each step are also available. These give great amount of flexibility to the user in choosing CPU, wall time, memory and queue for each step (and are passed along to the HPCC platform).
cpu_reservedmemory_reservednodeswalltimequeue
This is especially useful for genomics pipelines, since each step may use different amount of resources. For example, in a typical setup, if one step uses 16 cores these would be blocked and not used during processing of several other steps. Thus resulting in blockage and high cluster load (even when actual CPU usage may be low). Being able to tune them, makes this setup quite efficient.
Most cluster platforms accept these resource arguments. Essentially a file like this is used as a template, and variables defined in curly braces ( ex. {{{CPU}}} ) are filled up using the flow definition file.
Warning
If these (resource requirements) columns not included in the flow_def, their values should be explicitly defined in the submission template.
Here is an example of a typical flow_def file.
| jobname | sub_type | prev_jobs | dep_type | queue | memory_reserved | walltime | cpu_reserved | platform | jobid |
|---|---|---|---|---|---|---|---|---|---|
| sleep | scatter | none | none | short | 2000 | 1:00 | 1 | torque | 1 |
| create_tmp | scatter | sleep | serial | short | 2000 | 1:00 | 1 | torque | 2 |
| merge | serial | create_tmp | gather | short | 2000 | 1:00 | 1 | torque | 3 |
| size | serial | merge | serial | short | 2000 | 1:00 | 1 | torque | 4 |
2. Flow mat: A table with shell commands to run¶
This is also a tab separated table, with a minimum of three columns as defined below:
samplename: A grouping column. The table is split using this column and each subset is treated as a individual flow. This makes it very easy to process multiple samples using a single submission command.- If all the commands are for a single sample, one can just repeat a dummy name like sample1 all throughout.
jobname: This corresponds to the name of the step. This should match exactly with the jobname column in flow_def table defined above.cmd: A shell command to run. One can get quite creative here. These could be multiple shell commands separated by a;or&&, more on this here. Though to keep this clean you may just wrap a multi-line command into a script and just source the bash script from here.
Here is an example flow_mat.
| samplename | jobname | cmd |
|---|---|---|
| sample1 | sleep | sleep 10 && sleep 2;echo hello |
| sample1 | sleep | sleep 11 && sleep 8;echo hello |
| sample1 | sleep | sleep 11 && sleep 17;echo hello |
| sample1 | create_tmp | head -c 100000 /dev/urandom > sample1_tmp_1 |
| sample1 | create_tmp | head -c 100000 /dev/urandom > sample1_tmp_2 |
| sample1 | create_tmp | head -c 100000 /dev/urandom > sample1_tmp_3 |
| sample1 | merge | cat sample1_tmp_1 sample1_tmp_2 sample1_tmp_3 > sample1_merged |
| sample1 | size | du -sh sample1_merged; echo MY shell: $SHELL |
Example:¶
A —-> B —–> C —–> D
Consider an example with three steps A, B and C. A has 10 commands from A1 to A10, similarly B has 10 commands B1 through B10 and C has a single command, C1.
Consider another step D (with D1-D3), which comes after C.
Submission types¶
This refers to the sub_type column in flow definition.
scatter: submit all commands as parallel, independent jobs.- Submit A1 through A10 as independent jobs
serial: run these commands sequentially one after the other.- Wrap A1 through A10, into a single job.
Dependency types¶
This refers to the dep_type column in flow definition.
none: independent job.- Initial step A has no dependency
serial: one to one relationship with previous job.- B1 can start as soon as A1 completes.
gather: many to one, wait for all commands in previous job to finish then start the current step.- All jobs of B (1-10), need to complete before C1 is started
burst: one to many wait for the previous step which has one job and start processing all cmds in the current step.- D1 to D3 are started as soon as C1 finishes.
Relationships¶
Using the above submission and dependency types one can create several types of relationships between former and later jobs. Here are a few pipelines of relationships one may typically use.
Serial: one to one relationship¶
[scatter] —serial—> [scatter]
A is submitted as scatter, A1 through A10. Further B1, requires A1 to complete; B2 requires A2 and so on, but they need not wait for all of step A jobs to complete. Also B1 through B10 are independent of each other.
To set this up, A and B would have sub_type scatter and B would have dep_type as serial. Further, since A is an initial step its dep_type and prev_job would defined as none.
Gather: many to one relationship¶
[scatter] —gather—> [serial]
Since C is a single command which requires all steps of B to complete, intuitively it needs to gather pieces of data generated by B. In this case dep_type would be gather and sub_type type would be serial since it is a single command.
Burst: one to many relationship¶
[serial] —burst—> [scatter]
Further, D is a set of three commands (D1-D3), which need to wait for a single process (C1) to complete. They would be submitted as scatter after waiting on C in a burst type dependency.
In essence and example flow_def would look like as follows (with additional resource requirements not shown for brevity).
ex2def = as.flowdef(file.path(ex, "abcd.def"))
ex2mat = as.flowmat(file.path(ex, "abcd.tsv"))
fobj = suppressMessages(to_flow(x = ex2mat, def = ex2def))
kable(ex2def[, 1:4])
| jobname | sub_type | prev_jobs | dep_type |
|---|---|---|---|
| A | scatter | none | none |
| B | scatter | A | serial |
| C | serial | B | gather |
| D | scatter | C | burst |
plot_flow(fobj)
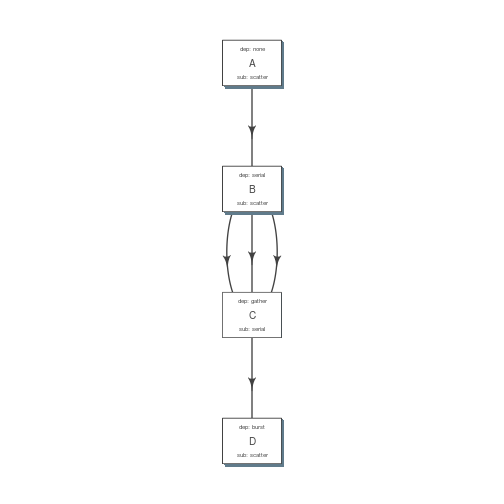
plot of chunk build_pipe_plt_abcd
There is a darker more prominent shadow to indicate scatter steps.
Passing of flow definition resource columns¶
The resource requirement columns of flow definition are passed along to the final (cluster) submission script.
The following table provides a mapping between the flow definition columns and variables in the submission template (pipelines below).
| flow_def_column | hpc_script_variable |
|---|---|
| nodes | NODES |
| cpu_reserved | CPU |
| memory_reserved | MEMORY |
| walltime | WALLTIME |
| extra_opts | EXTRA_OPTS |
| * | JOBNAME |
| * | STDOUT |
| * | CWD |
| * | DEPENDENCY |
| * | TRIGGER |
| ** | CMD |
*: These are generated on the fly **: This is gathered from flow_mat
Available Pipelines¶
Here are some of the available piplines along with their respective locations
| name | def | conf | pipe |
|---|---|---|---|
| sleep_pipe | sleep_pipe.def | NA | /home/travis/build/sahilseth/flowr/inst/pipelines/sleep_pipe.R |
Cluster Support¶
Support for several popular cluster platforms are built-in. There is a template, for each platform, which should would out of the box. Further, one may copy and edit them (and save to ~/flowr/conf) in case some changes are required. Templates from this folder (~/flowr/conf), would override defaults.
Here are links to latest templates on github:
Adding a new plaform involves a few steps, briefly we need to consider the following steps where changes would be neccesary.
- job submission: One needs to add a new template for the new platform. Several examples are available as described in the previous section.
- parsing job ids: flowr keeps a log of all submitted jobs, and also to pass them along as a dependency to subsequent jobs. This is taken care by the parse_jobids() function. Each job scheduler shows the jobs id, when you submit a job, but each shows it in a slightly different pattern. To accomodate this one can use regular expressions as described in the relevent section of the flowr config.
For example LSF may show a string such as:
Job <335508> is submitted to queue <transfer>.
jobid="Job <335508> is submitted to queue <transfer>."
set_opts(flow_parse_lsf = ".*(\<[0-9]*\>).* ")
parse_jobids(jobid, platform="lsf")
[1] "335508"
In this case 335508 was the job id and regex worked well !
- render dependency: After collecting job ids from previous jobs, flowr render them as a dependency for subsequent jobs. This is handled by render_dependency.PLATFORM functions.
- recognize new platform: Flowr needs to be made aware of the new platform, for this we need to add a new class using the platform name. This is essentially a wrapper around the job class
Essentially this requires us to add a new line like: setClass("torque", contains = "job").
- killing jobs: Just like submission flowr needs to know what command to use to kill jobs. This is defined in detect_kill_cmd function.
There are several job scheduling systems available and we try to support the major players. Adding support is quite easy if we have access to them. Your favourite not in the list? re-open this issue, with details on the platform: adding platforms
As of now we have tested this on the following clusters:
| Platform | command | status | queue.type |
|---|---|---|---|
| LSF 7 | bsub | Not tested | lsf |
| LSF 9.1 | bsub | Yes | lsf |
| Torque | qsub | Yes | torque |
| SGE | qsub | Beta | sge |
| SLURM | sbatch | under-dev | slurm |
*queue short-name used in flow
- PBS: wiki
- Torque: wiki
- MD Anderson
- University of Houston
- LSF wiki:
- SGE wiki
- A tutorial for Sun Grid Engine
- Another from JHSPH
- Dependecy info here
Example of building a pipeline¶
A pipeline consists of several pieces, namely, a function which generates a flowmat, a flowdef and optionally a text file with parameters and paths to tools used as part of the pipeline.
A R function which creates a flow mat, is a module. Further a module with a flow definition is a pipeline.
We beleive pipeline and modules may be interchangeble, in the sense that a smaller pipeline may be included as part of a larger pipeline. In flowr a module OR pipeline always returns a flowmat. The only differnce being, a pipeline also has a correspomding flow definition file. As such, creating a flow definition for a module enables flowr to run it, hence a module elevates, becoming a pipeline. This lets the user mix and match several modules/pipelines to create a customized larger pipeline(s).
Let us follow through an example, providing more details regarding this process. Here are a few examples of modules, three functions sleep, create_tmp and merge_size each returning a flowmat.
Define modules¶
#' @param x number of sleep commands
sleep <- function(x, samplename){
cmd = list(sleep = sprintf("sleep %s && sleep %s;echo 'hello'",
abs(round(rnorm(x)*10, 0)),
abs(round(rnorm(x)*10, 0))))
flowmat = to_flowmat(cmd, samplename)
return(list(flowmat = flowmat))
}
#' @param x number of tmp commands
create_tmp <- function(x, samplename){
## Create 100 temporary files
tmp = sprintf("%s_tmp_%s", samplename, 1:x)
cmd = list(create_tmp = sprintf("head -c 100000 /dev/urandom > %s", tmp))
## --- convert the list into a data.frame
flowmat = to_flowmat(cmd, samplename)
return(list(flowmat = flowmat, outfiles = tmp))
}
#' @param x vector of files to merge
merge_size <- function(x, samplename){
## Merge them according to samples, 10 each
mergedfile = paste0(samplename, "_merged")
cmd_merge <- sprintf("cat %s > %s",
paste(x, collapse = " "), ## input files
mergedfile)
## get the size of merged files
cmd_size = sprintf("du -sh %s; echo 'MY shell:' $SHELL", mergedfile)
cmd = list(merge = cmd_merge, size = cmd_size)
## --- convert the list into a data.frame
flowmat = to_flowmat(cmd, samplename)
return(list(flowmat = flowmat, outfiles = mergedfile))
}
We then define another function sleep_pipe which calls the above defined modules; fetches flowmat from each, creating a larger flowmat. This time we will define a flowdef for the sleep_pipe function, elevating its status from module to a pipeline.
Define the pipeline¶
#' @param x number of files to make
sleep_pipe <- function(x = 3, samplename = "samp1"){
## call the modules one by one...
out_sleep = sleep(x, samplename)
out_create_tmp = create_tmp(x, samplename)
out_merge_size = merge_size(out_create_tmp$outfiles, samplename)
## row bind all the commands
flowmat = rbind(out_sleep$flowmat,
out_create_tmp$flowmat,
out_merge_size$flowmat)
return(list(flowmat = flowmat, outfiles = out_merge_size$outfiles))
}
Generate a flowmat¶
Here is how the generated flowmat looks like.
out = sleep_pipe(x = 3, "sample1")
flowmat = out$flowmat
| samplename | jobname | cmd |
|---|---|---|
| sample1 | sleep | sleep 16 && sleep 17;echo ‘hello’ |
| sample1 | sleep | sleep 26 && sleep 8;echo ‘hello’ |
| sample1 | sleep | sleep 6 && sleep 22;echo ‘hello’ |
| sample1 | create_tmp | head -c 100000 /dev/urandom > sample1_tmp_1 |
| sample1 | create_tmp | head -c 100000 /dev/urandom > sample1_tmp_2 |
| sample1 | create_tmp | head -c 100000 /dev/urandom > sample1_tmp_3 |
| sample1 | merge | cat sample1_tmp_1 sample1_tmp_2 sample1_tmp_3 > sample1_merged |
| sample1 | size | du -sh sample1_merged; echo ‘MY shell:’ $SHELL |
Create flow definition¶
flowr enables us to quickly create a skeleton flow definition using a flowmat, which we can then alter to suit our needs. A handy function to_flowdef, accepts a flowmat and creates a flow definition. The default skeleton takes a very conservative approach, creating all submissions as serial and all dependencies as gather. This ensures robustness, compromising efficiency. Thus we will enable parallel process where possible, making this into a better pipeline.
Here is how it looks presently:
def = to_flowdef(flowmat)
## Creating a skeleton flow definition
## Following jobnames detected: sleep create_tmp merge size
## checking submission and dependency types...
plot_flow(suppressMessages(to_flow(flowmat, def)))
## checking submission and dependency types...
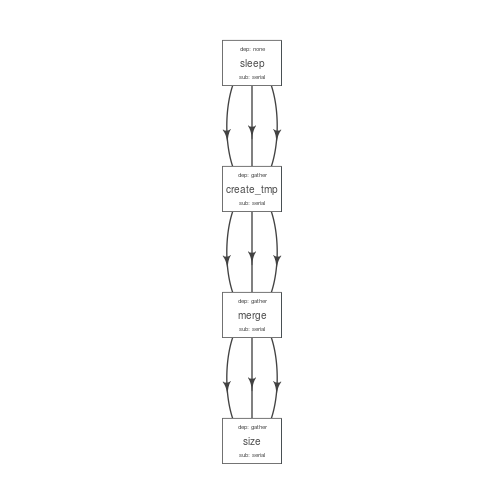
plot of chunk unnamed-chunk-18
After making the desired changes, the new pipeline looks better. Alternatively, one may write this to a file and make other desired changes in resource requirements.
Pipeline follows the following steps, with dependencies mentioned in ():
- multiple sleep commands would run in parallel (none, first step)
- For each sleep, create_tmp creates a tmp file (serial)
- All tmp files are merged; when all are complete (gather)
- Then we get size on the resulting file (serial)
def$sub_type = c("scatter", "scatter", "serial", "serial")
def$dep_type = c("none", "serial", "gather", "serial")
kable(def)
| jobname | sub_type | prev_jobs | dep_type | queue | memory_reserved | walltime | cpu_reserved | platform | jobid |
|---|---|---|---|---|---|---|---|---|---|
| sleep | scatter | none | none | short | 2000 | 1:00 | 1 | torque | 1 |
| create_tmp | scatter | sleep | serial | short | 2000 | 1:00 | 1 | torque | 2 |
| merge | serial | create_tmp | gather | short | 2000 | 1:00 | 1 | torque | 3 |
| size | serial | merge | serial | short | 2000 | 1:00 | 1 | torque | 4 |
plot_flow(suppressMessages(to_flow(flowmat, def)))
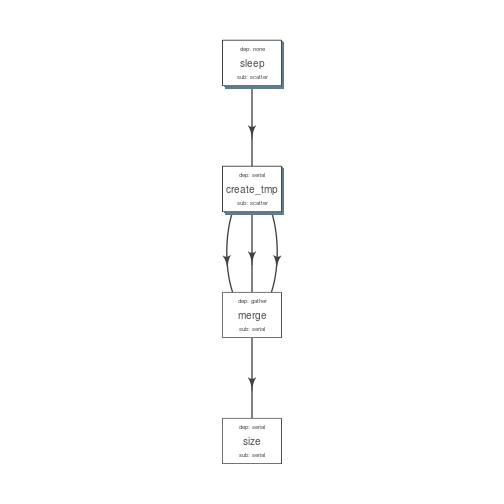
plot of chunk unnamed-chunk-20